Blogs & News
DRE-Enabled Clinical Operations
Clinical operations teams are responsible for designing, planning and physically running Phase I – IV clinical trials within the early stage drug development. Clinical operations (Clin Ops) also ensures patients receive the highest quality care. Beyond that, the clinical operation definition also encompasses adherence to regulatory requirements and standards. Within a sponsor company the ops department works to establish that all clinical staff and patients have what they need to work together well and see that the process is smooth.
Specifically, these teams can come into play during the drug development lifecycle. Once discovery and preclinical research are complete and the governing regulatory authorities give the go ahead to enter the human phase of testing (e.g., following IND approval), the clinical development process begins. At this stage (Phase 1 and beyond), clinical trials and volunteer studies occur to ensure the new drug candidate is safe for human use. Clin Ops play a vital role in making sure those trials are a success.
Getting Started
The study startup stage includes building a budget, designing a clinical trial protocol, and plenty of preclinical research. Clinical operations teams are responsible for designing, planning and physically running Phase I – IV clinical trials within the drug discovery industry. Depending on the size and type of clinical trial being conducted, clinical operations teams can be either small regional teams or large international groups of people. They are often made up of:
• Administrative roles or clinical trial administrators who support the day-to-day activities for the trial.
• Field-based monitors or clinical research associates who travel to different research units to ensure the trial is running correctly.
• Clinical study/project managers who manage the teams or running of the trial (usually on a regional or global level).
• Directorship roles where the person will have the sole responsibility for a trial.
Teams have a great deal of interaction with internal colleagues from other departments including clinical science, clinical quality assurance, data management, biostatistics and regulatory affairs to ensure that the data and information needed by these other departments is delivered so they can decide if a trial has been successful. Efficient coordination is an essential component of high functional Clin Ops teams. Table 1 below outlines the major functions of early stage clinical operations activities.
| Activity | Description |
|---|---|
| Study supplies and tracking | The supply of materials required for conducting a clinical trial which likely includes 24-hour collection and delivery of biological specimens (including infectious samples), investigational drugs, kits and other study materials and supplies, expertise/assistance |
| Safety monitoring and reporting | The goal of safety monitoring in clinical trials is to identify, evaluate, minimize and appropriately manage risks. In Europe, Risk Management Plans (RMPs) are required by the EMA as part of the drug approval process. |
| Subject randomization and progress | Randomization is a method of allocating subjects in a clinical trial to treatment groups such that every subject has an equal chance of receiving any one of the treatments or interventions. This enables researchers to isolate the causal effect of their treatment and make valid comparisons between groups. |
| EDC – data processing | Electronic Data Capture (EDC) Systems collect, store, and manage clinical data. Subsequent data processing creates the tables and listings used to communicate in process study results with the Drug Safety Monitoring Board (DSMB) and regulatory authorities. |
| Site monitoring | Clinical site monitoring is an element of the overall data and safety monitoring of clinical research. The purpose of clinical site monitoring is to ensure compliance with the protocol, Good Clinical Practice, Federal and local regulations, and institutional policies. |
| Regulatory compliance | Regulatory compliance is an organization’s adherence to laws, regulations, guidelines and specifications relevant to its business processes. Violations of regulatory compliance often result in legal punishment, including federal fines. For drug development sponsors in the US, the FDA is the relevant regulatory authority. |
There is an order to the activity of course and the effort requires a lot of coordination on behalf of Clinical Operations, Project Management, the clinical groups responsible for the science and the protocol content as well as Regulatory Affairs of course. Since clinical trials are experiments in humans, they must be conducted following established standards in order to protect the rights, safety and well-being of the participants. These standards include the International Conference on Harmonization Good Clinical Practice (ICH-GCP) Guidelines [Dixon 1998], International Ethical Guidelines for Biomedical Research Involving Human Subjects issued by the Council for International Organizations of Medical Sciences (CIOMS) [CIOMS 2016] and the ethical principles set forth in the Declaration of Helsinki [Goodyear 2007]. GCP is the “standard for the design, conduct, performance, monitoring, auditing, recording, analyses and reporting of clinical trials that provides assurance that the data and reported results are credible and accurate and that rights, integrity and confidentiality of trial subjects are protected” [Goodyear 2007].
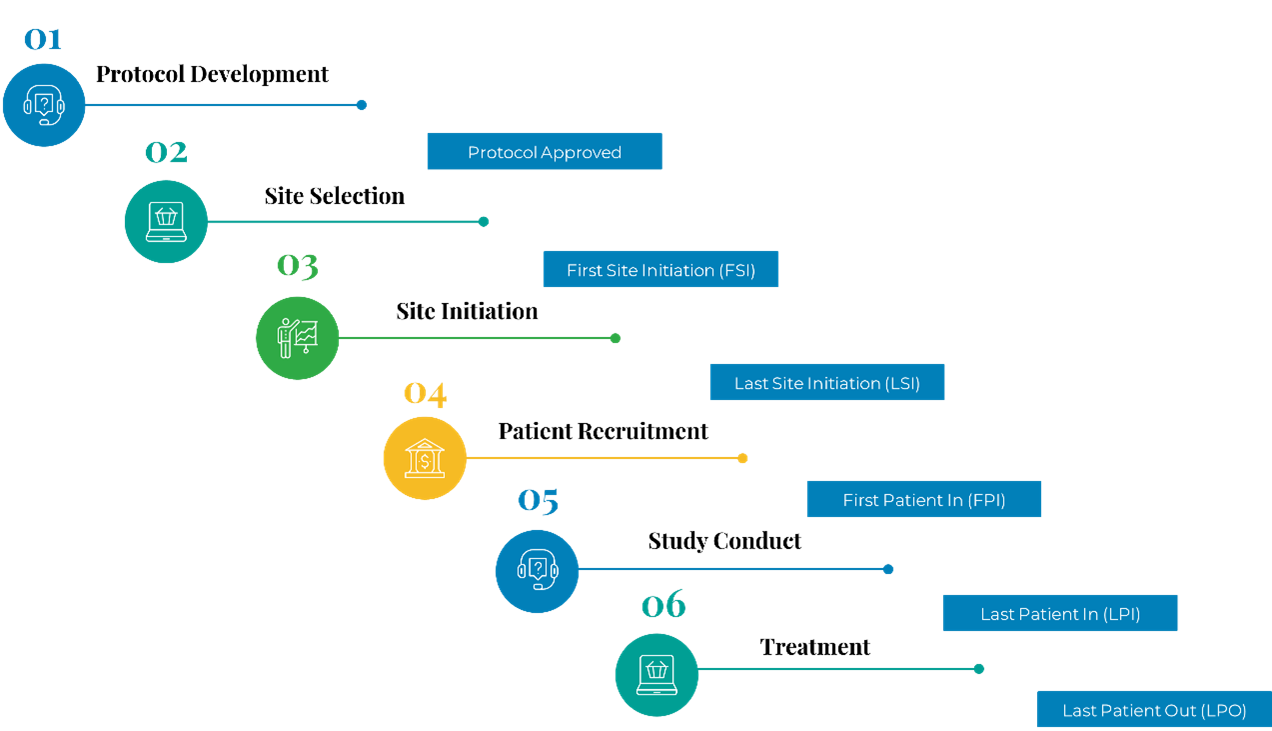
Figure 1. Generalized Clinical Operations Workflow with Key Milestones (Blue boxes)
In order to perform the tasks outlined in Figure 1, Clin Ops staff must interact with a diverse array of stakeholders as previously mentioned. In the past, such communications were primarily managed via phone calls and messages, faxing of documents (to the DSMB and Regulatory authorities), and FTP transfer of sensitive information. Much of the site initiation was completed via in-person site visits. Some electronic systems such as EDC and clinical trial management software [CTMS review 2023] did evolve, but these are often separate systems with unique administrators where only a select number of the aforementioned stakeholders interact. Likewise, each requires licensing and installation / validation / update maintenance from internal resources.
How the Digital Research Environment can help
The Aridhia DRE can support both home grown and commercial clinical operations solutions. More generally, the Digital Research Environment (DRE) is a secure end-to-end biomedical research facilitation platform, connecting data owners to researchers in a controlled and audited manner, ensuring data owners are always in control of how their data is used, who it can be used by and what it can be used for. For Clin Ops teams, the DRE can provide secure collaborative workspaces to work with approved data, with specialised tooling to meet their research, development and modelling requirements with data ingress and egress controls. This would enable secure and dynamic interactions with external DSMB members, study statisticians, and potentially external regulatory authorities if necessary.
Certain other clinical operational aspects of a trial can be facilitated with the DRE as well, such as the maintenance and compliance of study blinding and the communication with the drug safety monitoring board (DSMB). In both situations, secure and private communication with protocol-predefined roles can be managed with data and workspace services. Sponsors control the access and sharing of data based on an “airlocking mechanism” which is part of the workspace functionality. Specifically, a workspace with protocol-specified unblinded data and individuals can be maintained with study monitors and other clinical roles dealing only with accession numbers. Likewise, a secure workspace can be created with only the DSMB where data, results, and listings can be accessed per protocol and only for finite observation times (i.e., DSMB members are “invited” to access the workspace at certain times without access to other study-related data).
Trial By DRE
Recently, Aridhia scientists in collaboration with external SMEs have proposed a rationale for utilizing the DRE to support Platform Trials. With the manuscript in review we’ve articulated a manner by which individual PhRMA sponsors can collaborate in such trials while maintaining IP over sensitive data and yet still share enough to provide a means by which such trials can proceed with the appropriate governance in place (see Figure 2).
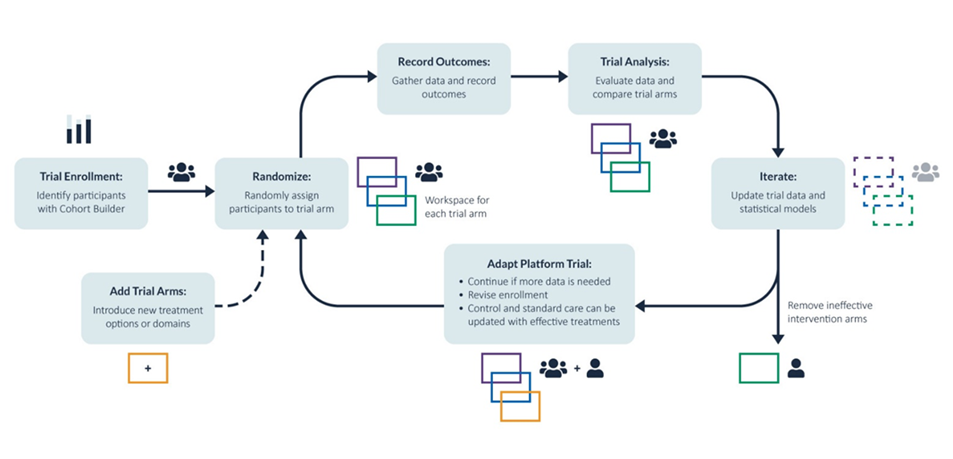
Figure 2. Schematic illustrating platform trial architecture and workflow within a digital research environment (DRE)
In reality, there is nothing magical about the platform trial architecture, and the DRE support outlined in the approach would support any trial design. Likewise, the gains in efficiency, while appreciated by the entire project team, would greatly benefit the work of the Clin Ops groups.
References:
- • Jekunen A. Decision-making in product portfolios of pharmaceutical research and development – managing streams of innovation in highly regulated markets. Drug Des Devel Ther. 2014; 8: 2009–2016.
- • Zeller C. Project Teams as Means of Restructuring Research and Development in the Pharmaceutical Industry. Regional Studies, Vol. 36.3, pp. 275–289, 2002.
- • Barrett JS, editor, Fundamentals of Drug Development, Wiley; 1st edition (September 7, 2022), ISBN-10: 1119691699, ISBN-13: 978-1119691693 (512 pages).