Blogs & News
RNA-seq: creating simple outputs from complex genetic data
As part of our recent stream of genomics work, we began to look at ways in which the complex data outputs associated with RNA-seq research could be effectively interpreted and presented in way that would make them aesthetically pleasing and easier to digest. This led to the creation of the RNA-seq mini-app.
Through the study of DNA we have learned that particular mutations in the genome can lead to increased susceptibility to disease or increased efficacy and toxicity of certain drugs. By studying RNA, we move closer to the mechanism of the cell and can gain greater understanding of why these mutations have the effects they do. This, in turn, leads to greater understanding of how to deal with the issues that can arise due to certain mutations and even predict what other effects they may have.
RNA is an important intermediate between the information hardcoded into the genome (genotype) and the functional output of a cell (phenotype). By capturing this dynamic information, RNA-sequencing enables investigation of how a cell or tissue’s function is regulated by genes. Genome expression has previously been measured on a genome scale using microarray, however RNA-seq allows de novo detection of transcripts and has a larger dynamic range.
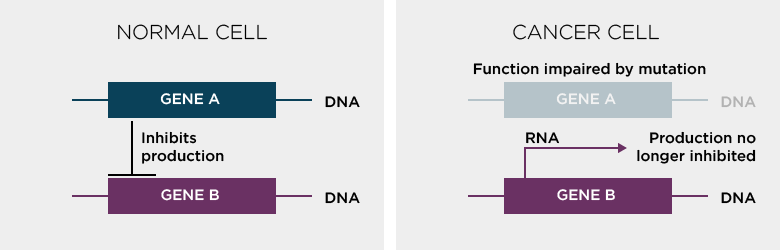
As an example imagine that, through DNA sequencing and subsequent analysis, a particular mutation has been found in Gene A that correlates with the occurrence of a disease. RNA-seq analysis identifies a different gene (Gene B) to be very highly expressed in samples which have the mutation in Gene A. Further analysis reveals that Gene A normally inhibits Gene B. When Gene A is mutated, high expression of Gene B is noted and this is what causes the disease. Product of gene B needs to be blocked for effective therapy. So although we initially found that Gene A was the root cause of the problem, an effective therapy might be to block Gene B.
We have created a mini-app within AnalytiXagility to showcase some of the analysis that can easily be performed and presented on RNA-seq data. The data we use is a normalised read counts file from a study on GEO, containing the read counts for both control and treated samples collected after 12 and 48 hours. The experiment used 3 biological replicates, producing 12 gene expression profiles to analyse. This data was uploaded to AnalytiXagility and some additional data pulled from Biomart (using the R package biomaRt) for annotation purposes.
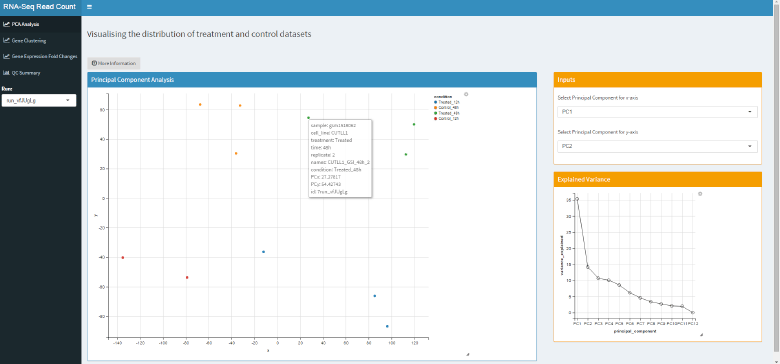
Principal Component Analysis
Principal component analysis is a method of dimensionality reduction and can be thought of heuristically as:
- Fit the smallest possible ellipsoid around our dataset and choose the longest axis as our first component.
- Proceed iteratively, choosing the next component as the next longest axis that is orthogonal to all the previous ones.
In the first tab of the mini-app we use principal component analysis to reduce the number of dimensions of our dataset so that we can plot as much information as possible onto two axes. From this we can visually assess the existence of clusters in our data.
We can see that with our 12 samples, we can only choose new components (non-arbitrarily) until we have 11 total. However, it is clear from the plot that the first two components do a pretty good job at separating the different experimental cases into groups.
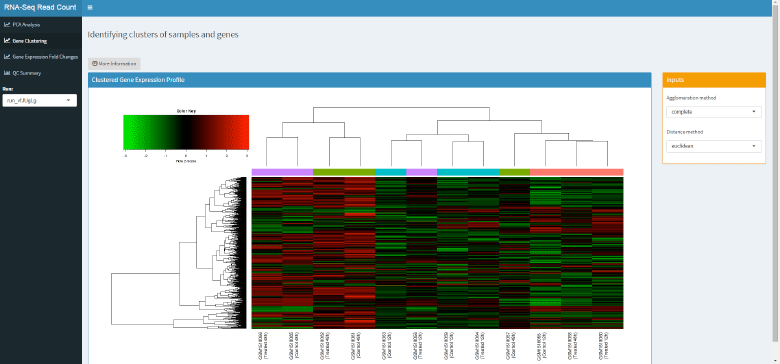
Expression Clustering
The second tab contains a heat map showing the expression profiles of the most variant genes across the study. Genes are represented by row, samples by columns, and the relative level of each gene represented by colour (red = high expression, green = low expression)
Columns and rows are clustered enabling the user to visualise groups of samples with similar expression profiles, and groups of genes which change across the data. The mini-app also allows the user to change the agglomeration method and the distance metric used to assess proximity.
Differential Expression Analysis
The third tab plots the results of a differential expression analysis, i.e. a comparison of the expression level of each gene between two groups of samples. Every point represents a gene, plotted by log2 fold change between sample groups and log2 mean expression level. Genes of interest are highlighted above or below a certain threshold log2 fold change (specified by the slider input). Each point can be hovered over to reveal gene specific information.
QC Summary
Finally, in the fourth tab, we show a basic QC summary from an RNA-seq pipeline where different runs can be selected and the outputs of the pipeline seen. In this example we present the results from checking the quality of read alignment using Hisat. Alternate runs can also be selected across the mini-app as a whole and the results of the previously described analyses shown for each. This shows that we could have the results of an RNA-seq pipeline being fed directly into AnalytiXagility and this mini-app (or similar) could be presented on top to quickly flag up interesting patterns or any issues with the results.
This really just scratches the surface of RNA-seq analysis shows that a range of different techniques are easy to implement in R and then present in a mini-app within AnalytiXagility. If you would like to find out more about how AnalytiXagility supports genomic research please get in touch.